Drug discovery with explainable artificial intelligence
Deep learning bears promise for drug discovery, including advanced image analysis, prediction of molecular structure and function, and automated generation of innovative chemical entities with bespoke properties. Despite the growing number of successful prospective applications, the underlying mathematical models often remain elusive to interpretation by the human mind. There is a demand for ‘explainable’ deep learning methods to address the need for a new narrative of the machine language of the molecular sciences. This Review summarizes the most prominent algorithmic concepts of explainable artificial intelligence, and forecasts future opportunities, potential applications as well as several remaining challenges. We also hope it encourages additional efforts towards the development and acceptance of explainable artificial intelligence techniques.
Similar content being viewed by others
Artificial intelligence for natural product drug discovery
Article 11 September 2023

Computational approaches streamlining drug discovery
Article 26 April 2023
Integrating QSAR modelling and deep learning in drug discovery: the emergence of deep QSAR
Article 08 December 2023
Main
Various concepts of ‘artificial intelligence’ (AI) have been successfully adopted for computer-assisted drug discovery in the past few years 1,2,3 . This advance is mostly owed to the ability of deep learning algorithms, that is, artificial neural networks with multiple processing layers, to model complex nonlinear input–output relationships, and perform pattern recognition and feature extraction from low-level data representations. Certain deep learning models have been shown to match or even exceed the performance of the familiar existing machine learning and quantitative structure–activity relationship (QSAR) methods for drug discovery 4,5,6 . Moreover, deep learning has boosted the potential and broadened the applicability of computer-assisted discovery, for example, in molecular design 7,8 , chemical synthesis planning 9,10 , protein structure prediction 11 and macromolecular target identification 12,13 .
The ability to capture intricate nonlinear relationships between input data (for example, chemical structure representations) and the associated output (for example, assay readout) often comes at the price of limited comprehensibility of the resulting model. While there have been efforts to explain QSARs in terms of algorithmic insights and molecular descriptor analysis 14,15,16,17,18,19 , deep neural network models notoriously elude immediate accessibility by the human mind 20 . In medicinal chemistry in particular, the availability of ‘rules of thumb’ correlating biological effects with physicochemical properties underscores the willingness, in certain situations, to sacrifice accuracy in favour of models that better fit human intuition 21,22,23 . Thus, blurring the lines between the ‘two QSARs’ 24 (that is, mechanistically interpretable versus highly accurate models) may be key to accelerated drug discovery with AI 25 .
Automated analysis of medical and chemical knowledge to extract and represent features in a human-intelligible format dates back to the 1990s 26,27 , but has been receiving increasing attention due to the re-emergence of neural networks in chemistry and healthcare. Given the current pace of AI in drug discovery and related fields, there will be an increased demand for methods that help us understand and interpret the underlying models. In an effort to mitigate the lack of interpretability of certain machine learning models, and to augment human reasoning and decision-making, 28 , attention has been drawn to explainable AI (XAI) approaches 29,30 .
Providing informative explanations alongside the mathematical models aims to (1) render the underlying decision-making process transparent (‘understandable’) 31 , (2) avoid correct predictions for the wrong reasons (the so-called clever Hans effect) 32 , (3) avert unfair biases or unethical discrimination 33 and (4) bridge the gap between the machine learning community and other scientific disciplines. In addition, effective XAI can help scientists navigate ‘cognitive valleys’ 28 , allowing them to hone their knowledge and beliefs on the investigated process 34 .
While the exact definition of XAI is still under debate 35 , in the authors’ opinion, several aspects of XAI are certainly desirable in drug design applications 29 :
- Transparency—knowing how the system reached a particular answer.
- Justification—elucidating why the answer provided by the model is acceptable.
- Informativeness—providing new information to human decision-makers.
- Uncertainty estimation—quantifying how reliable a prediction is.
In general, XAI-generated explanations can be categorized as global (that is, summarizing the relevance of input features in the model) or local (that is, based on individual predictions) 36 . Moreover, XAI can be dependent on the underlying model, or agnostic, which in turn affects the potential applicability of each method. In this framework, there is no one-fits-all XAI approach.
There are many domain-specific challenges for future AI-assisted drug discovery, such as the data representation fed to said approaches. In contrast to many other areas in which deep learning has been shown to excel, such as natural language processing and image recognition, there is no naturally applicable, complete, ‘raw’ molecular representation. After all, molecules—as scientists conceive them—are models themselves. Such an ‘inductive’ approach, which builds higher-order (for example, deep learning) models from lower-order ones (for example, molecular representations or descriptors based on observational statements) is therefore philosophically debatable 37 . The choice of the molecular ‘representation model’ becomes a limiting factor of the explainability and performance of the resulting AI model—as it determines of the content, type and interpretability of the chemical information retained (for example, pharmacophores, physicochemical properties, functional groups).
Drug design is not straightforward. It distinguishes itself from clear-cut engineering by the presence of error, nonlinearity and seemingly random events 38 . We have to concede our incomplete understanding of molecular pathology and our inability to formulate infallible mathematical models of drug action and corresponding explanations. In this context, XAI bears the potential to augment human intuition and skills for designing novel bioactive compounds with desired properties.
Designing new drugs epitomizes in the question whether pharmacological activity (‘function’) can be deduced from the molecular structure, and which elements of such structure are relevant. Multi-objective design poses additional challenges and sometimes ill-posed problems, resulting in molecular structures that too often represent compromise solutions. The practical approach aims to limit the number of syntheses and assays needed to find and optimize new hit and lead compounds, especially when elaborate and expensive tests are performed. XAI-assisted drug design is expected to help overcome some of these issues, by allowing to take informed action while simultaneously considering medicinal chemistry knowledge, model logic and awareness on the system’s limitations 39 . XAI will foster the collaboration between medicinal chemists, chemoinformaticians and data scientists 40,41 . In fact, XAI already enables the mechanistic interpretation of drug action 42,43 , and contributes to drug safety enhancement, as well as organic synthesis planning 9,44 . If successful in the long run, XAI will provide fundamental support in the analysis and interpretation of increasingly more complex chemical data, as well as in the formulation of new pharmacological hypotheses, while avoiding human bias 45,46 . Pressing drug discovery challenges such as the coronavirus pandemic might boost the development of application-tailored XAI approaches, to promptly respond to specific scientific questions related to human biology and pathophysiology.
The field of XAI is still in its infancy but moving forward at a fast pace, and we expect an increase of its relevance in the years to come. In this Review, we aim to provide a comprehensive overview of recent XAI research, highlighting its benefits, limitations and future opportunities for drug discovery. In what follows, after providing an introduction to the most relevant XAI methods structured into conceptual categories, the existing and some of the potential applications to drug discovery are presented. Finally, we discuss the limitations of contemporary XAI and point to the potential methodological improvements needed to foster practical applicability of these techniques to pharmaceutical research.
A glossary of selected terms is provided in Box 1.
Box 1 Glossary of selected terms
Active learning. Field of machine learning in which an underlying model can query an oracle (for example, an expert or any other information source) in an active manner to label new data points with the goal of learning a task more efficiently.
Activity cliff. A small structural modification of a molecule that results in a marked change in its bioactivity.
Ensemble approach. Combination of the predictions of multiple base models with the goal to obtain a single one with improved overall performance metrics.
Cytochrome P450. Superfamily of structurally diverse metabolic enzymes, accounting for about 75% of the total drug metabolism in the human body.
Fragment-based virtual screening. Computational approach aimed to obtain promising hit or lead compounds based on the presence of specified molecular fragments (for example, molecular substructures known to possess or convey a certain desired biological activity).
Functional group. Part of a molecule that may be involved in characteristic chemical reactions or molecular interactions.
Gaussian process. Supervised, Bayesian-inspired machine learning model that naturally handles uncertainty estimation over its predictions. It does so by inducing a prior over functions with a covariance function that measures similarity among the inputs. Gaussian process models are often used for solving regression tasks.
Hit-to-lead optimization. Early stage of the drug discovery process in which the initial ‘hits’ (that is, molecules with a desired activity) undergo a filtering and optimization process to select the most promising ones (‘leads’).
Lead optimization. Process by which the potency, selectivity and pharmacokinetic parameters of a compound (‘lead structure’) are improved to obtain a drug candidate. This optimization usually involves several design–make–test cycles.
Metabolism. Biochemical reactions that transform and remove endogenous and exogenous compounds from an organism.
Molecular descriptor. Numerical representation of molecular properties and/or structural features, generated by predefined algorithmic rules.
Molecular graph. Mathematical representation of the molecular topology, with nodes and edges representing atoms and chemical bonds, respectively.
Pharmacophore. The set of molecular features that are necessary for the specific interaction of a ligand with a biological receptor.
SMILES. String-based representation of a two-dimensional molecular structure in terms of its atom types, bond types and molecular connectivity.
Structural alert. Functional group and/or molecular substructure empirically linked to adverse properties, for example, compound toxicity or unwanted reactivity.
QSAR model. ‘Quantitative structure–activity relationship’ approaches are methodologies for predicting the physicochemical or biological properties of chemicals as a function of their molecular descriptors.
State of the art and future directions
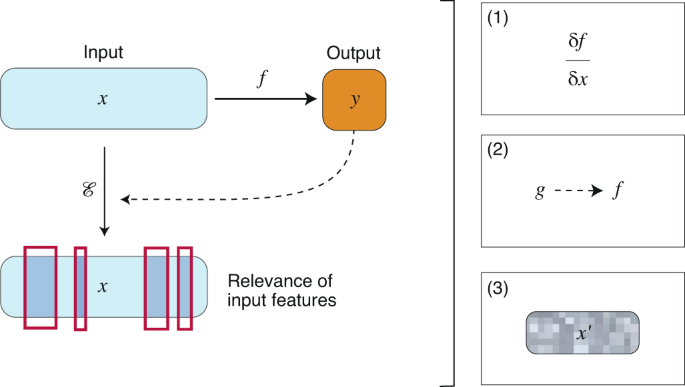
This section aims to provide a concise overview of modern XAI approaches, and exemplify their use in computer vision, natural-language processing and discrete mathematics. We then highlight selected case studies in drug discovery and propose potential future areas and research directions of XAI in drug discovery. A summary of the methodologies and their goals, along with reported applications is provided in Table 1. In what follows, without loss of generality, f will denote a model (in most cases a neural network); \(x \in >\) will be used to denote the set of features describing a given instance, which are used by f to make a prediction \(y \in >\) .
Feature attribution methods have been the most used XAI family of techniques for ligand- and structure-based drug discovery in the past few years. For instance, McCloskey et al. 66 employed gradient-based attribution 47 to detect ligand pharmacophores relevant for binding. The study showed that, despite good performance of the models on held-out data, these still can learn spurious correlations 66 . Pope et al. 67 adapted gradient-based feature attribution 68,69 for the identification of relevant functional groups in adverse effect prediction 70 . Recently, SHAP 52 was used to interpret relevant features for compound potency and multitarget activity prediction 71 . Hochuli et al. 72 compared several feature attribution methodologies, showing how the visualization of attributions assists in the parsing and interpretation of protein-ligand scoring with three-dimensional convolutional neural networks 73,74 .
It should be noted that the interpretability of feature attribution methods is limited by the original set of features (model input). Particularly in drug discovery, the interpretability is often hampered by the use of complex or ‘opaque’ input molecular descriptors 75 . When making use of feature attribution approaches, it is advisable to choose comprehensible molecular descriptors or representations for model construction (Box 2). Recently, architectures borrowed from the natural language processing field, such as long short-term memory networks 76 and transformers 77 , have been used as feature attribution techniques to identify portions of simplified molecular input line entry systems (SMILES) 78 strings that are relevant for bioactivity or physicochemical properties 79,80 . These approaches constitute a first attempt to bridge the gap between the deep learning and medicinal chemistry communities, by relying on representations (atom and bond types, and molecular connectivity 78 ) that bear direct chemical meaning and need no posterior descriptor-to-molecule decoding.
Box 2 XAI applied to cytochrome P450-mediated metabolism
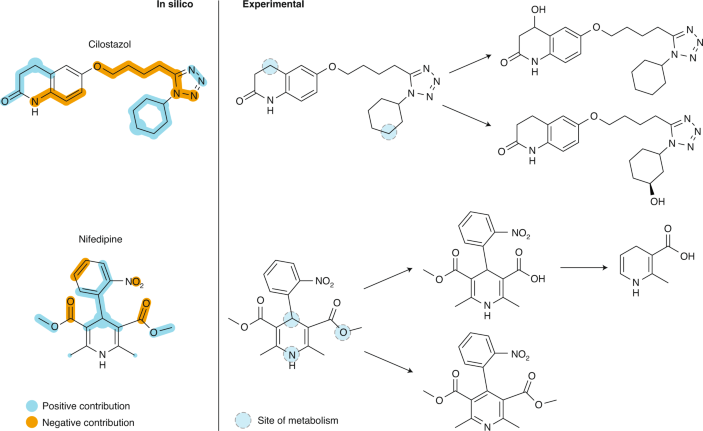
This worked example showcases XAI that provides a graphical explanation in terms of molecular motifs that are considered relevant by a neural network model predicting drug interaction with cytochrome P450 (3A4 isoform, CYP3A4). The integrated gradients feature attribution method 47 was combined with a graph convolutional neural network for predicting drug–CYP3A4 interaction. This network model was trained with a publicly available set of CYP3A4 substrates and inhibitors 169 . The figure shows the results obtained for two drugs that are metabolized predominantly by CYP3A4, namely the phosphodiesterase A inhibitor (antiplatelet agent) cilostazol and nifedipine, an L-type calcium channel blocker.
The structural features for CYP3A4–compound interaction suggested by the XAI method are highlighted in colour (left panel ‘in silico’: blue, positive contribution to interaction with CYP3A4; orange, negative contribution to interaction; spot size indicates the feature relevance of the respective atom). The main sites of metabolism (dashed circles) and the known metabolites 170,171,172 are shown in the right panel (‘experimental’). Apparently, the XAI captured the chemical substructures involved in CYP3A4-mediated biotransformation and most of the known sites of metabolism. Additional generic features related to metabolism were identified, that is, (1) the tetrazole moiety and the secondary amino group in cilostazol, which are known to increase metabolic stability (left panel: orange, negative contribution to the CYP3A4–cilostazol interaction), and (2) metabolically labile groups, such as methyl and ester groups (left panel: blue, positive contribution to the CYP3A4–nifedipine interaction).
Instance-based approaches
Instance-based approaches compute a subset of relevant features (instances) that must be present to retain (or absent to change) the prediction of a given model (Fig. 2). An instance can be real (that is, drawn from the set of data) or generated for the purposes of the method. Instance-based approaches have been argued to provide ‘natural’ model interpretations for humans, because they resemble counterfactual reasoning (that is, producing alternative sets of action to achieve a similar or different result) 81 .
- Anchor algorithms 82 offer model-agnostic interpretable explanations of classifier models. They compute a subset of if-then rules based on one or more features that represent conditions to sufficiently guarantee a certain class prediction. In contrast to many other local explanation methods 53 , anchors therefore explicitly model the ‘coverage’ of an explanation. Formally, an anchor A is defined as a set of rules such that, given a set of features x from a sample, they return A(x) = 1 if said rules are met, while guaranteeing the desired predicted class from f with a certain probability τ:
$$_>\left( \right)>\left[ <1_> \right] \ge \tau ,$$
where \(>\left( \right)\) is defined as the conditional distribution on samples where anchor A applies. This methodology has successfully been applied in several tasks such as image recognition, text classification and visual question answering 82 .
Counterfactual instance search. Given a classifier model f and an original data point x, counterfactual instance search 83 aims to find examples x′ (1) that are as close to x as possible and (2) for which the classifier produces a different class label from the label assigned to x. In other words, a counterfactual describes small feature changes in sample x such that it is classified differently by f. The search for the set of instances x′ may be cast into an optimization problem:
$$\mathop <<<\mathrm>>>\limits_ \mathop <<<\mathrm>>>\limits_\lambda \left( \right)^2 + \lambda L_1\left( \right),$$
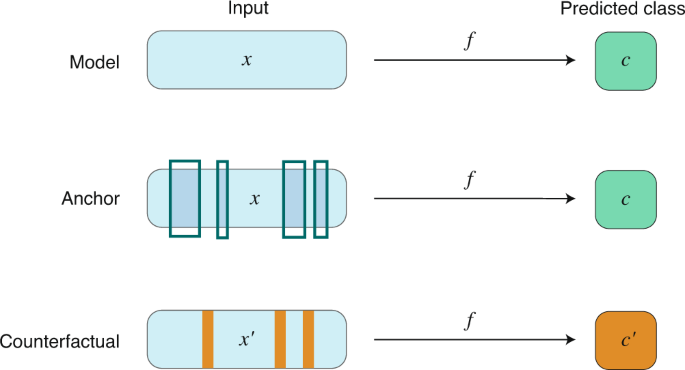
In drug discovery, instance-based approaches can be valuable to enhance model transparency, by highlighting what molecular features need to be either present or absent to guarantee or change the model prediction. In addition, counterfactual reasoning further promotes informativeness, by exposing potential new information about both the model and the underlying training data for human decision-makers (for example, organic and medicinal chemists).
To the best of our knowledge, instance-based approaches have yet to be applied to drug discovery. In the authors’ opinion, they bear promise in several areas of de novo molecular design, such as (1) activity cliff prediction, as they can help identify small structural variations in molecules that cause large bioactivity changes, (2) fragment-based virtual screening, by highlighting a minimal subset of atoms responsible for a given observed activity, and (3) hit-to-lead optimization, by helping identify the minimal set of structural changes required to improve one or more biological or physicochemical properties.
Graph-convolution-based methods
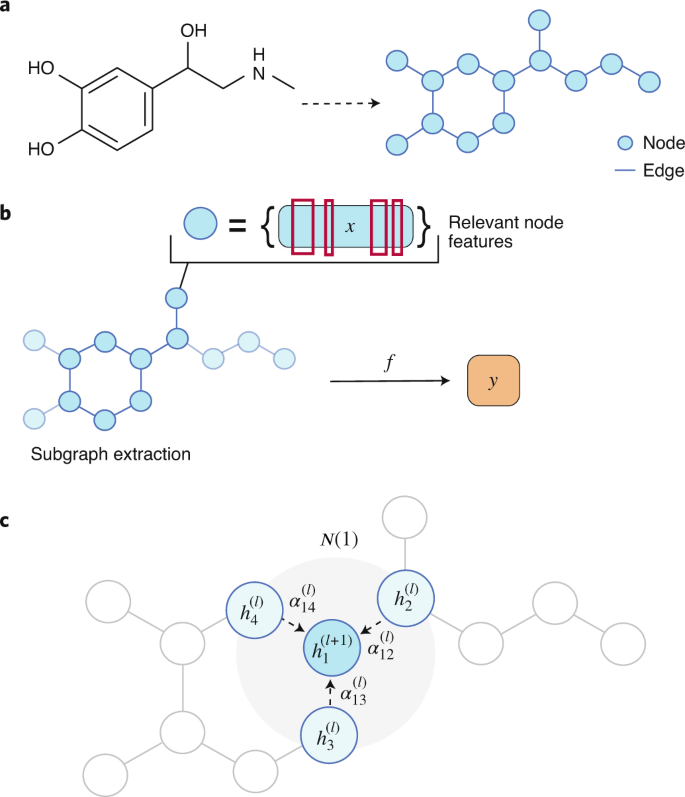
Molecular graphs are a natural mathematical representation of molecular topology, with nodes and edges representing atoms and chemical bonds, respectively (Fig. 3a) 75 . Their usage has been commonplace in chemoinformatics and mathematical chemistry since the late 1970s 88,89 . Thus, it does not come as a surprise in these fields to witness the increasing application of novel graph convolution neural networks 90 , which formally fall under the umbrella of neural message-passing algorithms 91 . Generally speaking, convolution refers to a mathematical operation on two functions that produces a third function expressing how the shape of one is modified by the other. This concept is widely used in convolutional neural networks for image analysis. Graph convolutions naturally extend the convolution operation typically used in computer vision 92 or in natural language processing 93 applications to arbitrarily sized graphs. In the context of drug discovery, graph convolutions have been applied to molecular property prediction 94,95 and in generative models for de novo drug design 96 .
Exploring the interpretability of models trained with graph convolution architectures is currently a particularly active research topic 97 . For the purpose of this review, XAI methods based on graph convolution are grouped into the following two categories.
Subgraph identification approaches aim to identify one or more parts of a graph that are responsible for a given prediction (Fig. 3b). GNNExplainer 98 is a model-agnostic example of this category, and provides explanations for any graph-based machine learning task. Given an individual input graph, GNNExplainer identifies a connected subgraph structure, as well a set of node-level features that are relevant for a particular prediction. The method can also provide such explanations for a group of data points belonging to the same class. GNNExplainer is formulated as an optimization problem, where a mutual information objective between the prediction of a graph neural network and the distribution of feasible subgraphs is maximized. Mathematically, given a node v, the goal is to identify a subgraph \(G_<\mathrm> \subseteq G\) with associated features \(X_<\mathrm> = \left\< >> \right\>\) that are relevant in explaining a target prediction \(\hat y \in Y\) via a mutual information measure MI:
$$h_i^ <\left( \right)> = \sigma \left( <\mathop <\sum>\nolimits_>\left( i \right)> ^lW^<\left( l \right)>h_j^<\left( l \right)>> > \right),$$
where \(>\left( i \right)\) is the set of topological neighbours of node i with a one-edge distance, \(\alpha _^l\) are learned attention coefficients over those neighbours, σ is a nonlinear activation function and W (l) is a learnable feature matrix for layer l. The main difference between this approach and a standard graph convolution update is that, in the latter, attention coefficients are replaced by a fixed normalization constant \(c_ = \sqrt <\left| <>\left( i \right)> \right|> \sqrt <\left| <>\left( j \right)> \right|>\) .
Methods based on graph convolution represent a powerful tool in drug discovery due to their immediate and natural connection with representations that are intuitive to chemists (that is, molecular graphs and subgraphs). In addition, the possibility to highlight atoms that are relevant towards a particular prediction, when combined with mechanistic knowledge, can improve both a model justification (that is, to elucidate if a provided answer is acceptable) and its informativeness on the underlying biological and chemical processes.
In particular, GNNExplainer was tested on a set of molecules labelled for their mutagenic effect on Salmonella typhimurium 100 , and identified several known mutagenic functional groups (that is, certain aromatic and heteroaromatic rings and amino/nitro groups 100 ) as relevant. A recent study 41 describes how the interpretation of filters in message-passing networks can lead to the identification of relevant pharmacophore- and toxicophore-like substructures, showing consistent findings with literature reports. Gradient-based feature attribution techniques, such as integrated gradients 47 , were used in conjunction with graph convolutional networks to analyse retrosynthetic reaction predictions and highlight the atoms involved in each reaction step 101 . Attention-based graph convolutional neural networks have also been used for the prediction of solubility, polarity, synthetic accessibility and photovoltaic efficiency, among other properties 102,103 , leading to the identification of relevant molecular substructures for the target properties. Finally, attention-based graph architectures have also been used in chemical reactivity prediction 104 , pointing to structural motifs that are consistent with a chemist’s intuition in the identification of suitable reaction partners and activating reagents.
Due to their intuitive connection with the two-dimensional representation of molecules, graph-convolution-based XAI bears the potential of being applicable to several other common modelling tasks in drug discovery. In the authors’ opinion, XAI for graph convolution might be mostly beneficial to applications aimed at finding relevant molecular motifs, for example, structural alert identification and site of reactivity or metabolism prediction.
Self-explaining approaches
The XAI methods introduced so far produce a posteriori explanations of deep learning models. Although such post hoc interpretations have been shown to be useful, some argue that, ideally, XAI methods, should automatically offer human-interpretable explanation alongside their predictions 105 . Such approaches (herein referred to as ‘self-explaining’) would promote verification and error analysis, and be directly linkable with domain knowledge 106 . While the term self-explaining has been coined to refer to a specific neural network architecture—self-explaining neural networks 106 , described below—in this Review, the term is used in a broader sense, to identify methods that feature interpretability as a central part of their design. Self-explaining XAI approaches can be grouped into the following categories.
- Prototype-based reasoning refers to the task of forecasting future events (that is, novel samples) based on particularly informative known data points. Usually, this is done by identifying prototypes, that is, representative samples, which are adapted (or used directly) to make a prediction. These methods are motivated by the fact that predictions based on individual, previously seen examples mimic human decision-making 107 . The Bayesian case model 108 is a pre-deep-learning approach that constitutes a general framework for such prototype-based reasoning. A Bayesian case model learns to identify observations that best represent clusters in a dataset (that is, prototypes), along with a set of defining features for that cluster. Joint inference is performed on cluster labels, prototypes and extracted relevant features, thereby providing interpretability without sacrificing classification accuracy 108 . Recently, Li et al. 109 developed a neural network architecture composed of an autoencoder and a therein named ‘prototype layer’, whose units store a learnable weight vector representing an encoded training input. Distances between the encoded latent space of new inputs and the learned prototypes are then used as part of the prediction process. This approach was later expanded by Chen et al. 110 to convolutional neural networks for computer vision tasks.
- Self-explaining neural networks 106 aim to associate input or latent features with semantic concepts. They jointly learn a class prediction and generate explanations using a feature-to-concept mapping. Such a network model consists of (1) a subnetwork that maps raw inputs into a predefined set of explainable concepts, (2) a parameterizer that obtains coefficients for each individual explainable concept and (3) an aggregation function that combines the output of the previous two components to produce the final class prediction.
- Human-interpretable concept learning refers to the task of learning a class of concepts, that is, high-level combinations of knowledge elements 111 , from data, aiming to achieve human-like generalization ability. The Bayesian programme learning approach 112 was proposed with the goal of learning visual concepts in computer vision tasks. Such concepts were represented as probabilistic programmes expressed as structured procedures in an abstract description language 113 . The model then composes more complex programmes using the elements of previously learned ones using a Bayesian criterion. This approach has been shown to reach human-like performance in one-shot learning tasks 114,115 .
- Testing with concept activation vectors 116 computes the directional derivatives of the activations of a layer with respect to its input, towards the direction of a concept. Such derivatives quantify the degree to which the latter is relevant for a particular classification (for example, how important the concept ‘stripes’ is for the prediction of the class ‘zebra’). It does so by considering the mathematical association between the internal state of a machine learning model—seen as a vector space Em spanned by basis vectors em that correspond to neural activations—and human-interpretable activations residing in a different vector space Eh spanned by basis vectors eh. A linear function is computed that translates between these vector spaces (g: Em → Eh). The association is achieved by defining a vector in the direction of the values of a concept’s set of examples, and then training a linear classifier between those and random counterexamples, to finally take the vector orthogonal to the decision boundary.
- Natural language explanation generation. Deep networks can be designed to generate human-understandable explanations in a supervised manner 117 . In addition to minimizing the loss of the main modelling task, several approaches synthesize a sentence using natural language that explains the decision performed by the model, by simultaneously training generators on large datasets of human-written explanations. This approach has been applied to generate explanations that are both image and class relevant 118 . Another prominent application is visual question answering 119 . To obtain meaningful explanations, however, this approach requires a substantial amount of human-curated explanations for training, and might, thus, find limited applicability in drug discovery tasks.
Self-explaining methods possess several desirable aspects of XAI, but in particular we highlight their intrinsic transparency. By incorporating human-interpretable explanations at the core of their design, they avoid the common need of a post hoc interpretation methodology. The produced human-intelligible explanations might also provide natural insights on the justification of the provided predictions.
To the best of our knowledge, self-explaining deep learning has not been applied to chemistry or drug design yet. Including interpretability by design could help bridge the gap between machine representation and the human understanding of many types of problems in drug discovery. For instance, prototype-reasoning bears promise in the modelling of heterogeneous sets of chemicals with different modes of action, allowing the preservation of both mechanistic interpretability and predictive accuracy. Explanation generation (either concept or text based) is another potential solution to include human-like reasoning and domain knowledge in the model building task. In particular, explanation-generation approaches might be applicable to certain decision-making processes, such as the replacement of animal testing and in vitro to in vivo extrapolation, where human-understandable generated explanations constitute a crucial element.
Uncertainty estimation
Uncertainty estimation, that is, the quantification of errors in a prediction, constitutes another approach to model interpretation. While some machine learning algorithms, such as Gaussian processes 120 , provide built-in uncertainty estimation, deep neural networks are known for being poor at quantifying uncertainty 121 . This is one of the reasons why several efforts have been devoted to specifically quantify uncertainty in neural network-based predictions. Uncertainty estimation methods can be grouped into the following categories.
Ensemble approaches. Model ensembles improve the overall prediction quality and have become a standard for uncertainty estimates 122 . Deep ensemble averaging 123 is based on m identical neural network models that are trained on the same data and with a different initialization. The final prediction is obtained by aggregating the predictions of all models (for example, by averaging), while an uncertainty estimate can be obtained from the respective variance (Fig. 4a). Similarly, the sets of data on which these models are trained can be generated via bootstrap re-sampling 124 . A disadvantage of this approach is its computational demand, as the underlying methods build on m independently trained models. Snapshot ensembling 125 aims to overcome this limitation by periodically storing model states (that is, model parameters) along the training optimization path. These model ‘snapshots’ can be then used for constructing the ensemble.
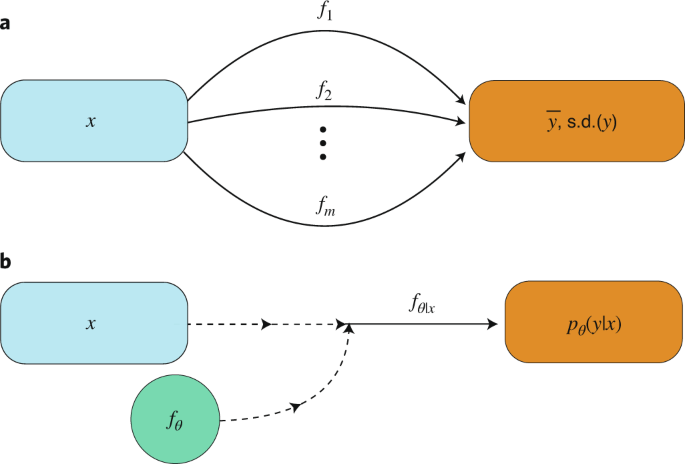
Uncertainty is omnipresent in the natural sciences, and errors can arise from several sources. The methods described in this Review mainly address the epistemic error, that is, uncertainty in the model and hyperparameter choice. However, the aleatoric error, that is, the intrinsic randomness related to the inherent noise in experimental data, is independent from in silico modelling. It should be noted that this distinction of error types is usually not taken into consideration in practice because these two types of error are often inseparable. Accurately quantifying both types of error, however, could potentially increase the value of the information provided to medicinal chemists in active learning cycles, and facilitate decision-making during the compound optimization process 74 .
Uncertainty estimation approaches have been successfully implemented in drug discovery applications 142 , mostly in traditional QSAR modelling, either by the use of models that naturally handle uncertainty 143 or post hoc methods 144,145 . Attention has recently been drawn towards the development of uncertainty-aware deep learning applications in the field. Snapshot ensembling was applied to model 24 bioactivity datasets 146 , showing that it performs on par with random forest and neural network ensembles, and also leads to narrower confidence intervals. Schwaller et al. 147 proposed a transformer model 77 for the task of forward chemical reaction prediction. This approach implements uncertainty estimation by computing the product of the probabilities of all predicted tokens in a SMILES sequence representing a molecule. Zhang et al. 148 have recently proposed a Bayesian treatment of a semi-supervised graph neural network for uncertainty-calibrated predictions of molecular properties, such as the melting point and aqueous solubility. Their results suggest that this approach can efficiently drive an active learning cycle, particularly in the low-data regime—by choosing those molecules with the largest estimated epistemic uncertainty. Importantly, a recent comparison of several uncertainty estimation methods for physicochemical property prediction showed that none of the methods systematically outperformed all others 149 .
Often, uncertainty estimation methods are applied alongside models that are difficult to interpret, due to the utilized algorithms, molecular descriptors or a combination of both. Importantly, however, uncertainty estimation alone does not necessarily avert several known issues of deep learning, such as a model producing the right answer for unrelated or wrong reasons or highly reliable but wrong predictions 31,32 . Thus, enriching uncertainty estimation with concepts of transparency or justification remains a fundamental area of research to maximize the reliability and effectiveness of XAI in drug discovery.
Available software
Given the attention deep learning applications are currently receiving, several software tools have been developed to facilitate model interpretation. A prominent example is Captum 150 , an extension of the PyTorch 151 deep learning and automatic differentiation package that provides support for most of the feature attribution techniques described in this work. Another popular package is Alibi 152 , which provides instance-specific explanations for certain models trained with the scikit-learn 153 or TensorFlow 154 packages. Some of the explanation methods implemented include anchors, contrastive explanations and counterfactual instances.
Conclusions and outlook
In the context of drug discovery, full comprehensibility of deep learning models may be hard to achieve 38 , although the provided predictions can still prove useful to the practitioner. When striving for interpretations that match the human intuition, it will be crucial to carefully devise a set of control experiments to validate the machine-driven hypotheses and increase their reliability and objectivity 40 .
Current XAI also faces technical challenges, given the multiplicity of possible explanations and methods applicable to a given task 155 . Most approaches do not come as readily usable, ‘out-of-the-box’ solutions, but need to be tailored to each individual application. In addition, profound knowledge of the problem domain is crucial to identify which model decisions demand further explanations, which type of answers are meaningful to the user and which are instead trivial or expected 156 . For human decision-making, the explanations generated with XAI have to be non-trivial, non-artificial and sufficiently informative for the respective scientific community. At least for the time being, finding such solutions will require the collaborative effort of deep-learning experts, chemoinformaticians and data scientists, chemists, biologists and other domain experts, to ensure that XAI methods serve their intended purpose and deliver reliable answers.
It will be of particular importance to further explore the opportunities and limitations of the established chemical language for representing the decision space of these models. One step forward is to build on interpretable ‘low level’ molecular representations that have direct meaning for chemists and are suited for machine learning (for example, SMILES strings 157,158 , amino acid sequences 159,160 and spatial three-dimensional voxelized representations 73,161 ). Many recent studies rely on well-established molecular descriptors, such as hashed binary fingerprints 162,163 and topochemical and geometrical descriptors 164,165 , which capture structural features defined a priori. Often, molecular descriptors, while being relevant for subsequent modelling, capture intricate chemical information. Consequently, when striving for XAI, there is an understandable tendency to employ molecular representations that can be more easily rationalized in terms of the known language of chemistry. Model interpretability depends on both the chosen molecular representation and the chosen machine learning approach 40 . With that in mind, the development of novel interpretable molecular representations for deep learning will constitute a critical area of research for the years to come, including the development of self-explaining approaches to overcome the hurdles of non-interpretable but information-rich descriptors, by providing human-like explanations alongside sufficiently accurate predictions.
Due to the current lack of methods comprising all of the outlined desirable features of XAI (transparency, justification, informativeness and uncertainty estimation), a major role in the short and mid term will be played by consensus (jury) approaches that combine the strengths of individual (X)AI approaches and increase model reliability. In the long run, jury XAI approaches—by relying on different algorithms and molecular representations—will constitute a way to provide multifaceted vantage points of the modelled biochemical process. Most of the deep learning models in drug discovery currently do not consider applicability domain restrictions 166,167 , that is, the region of chemical space where statistical learning assumptions are met. These restrictions should, in the authors’ opinion, be considered an integral element of XAI, as their assessment and a rigorous evaluation of model accuracy has proven to be more relevant for decision-making than the modelling approach itself 168 . Knowing when to apply which particular model will probably help address the problem of high confidence of deep learning models on wrong predictions 121 and avoid unnecessary extrapolations at the same time. Along those lines, in time- and cost-sensitive scenarios, such as drug discovery, deep learning practitioners have the responsibility to cautiously inspect and interpret the predictions derived from their modelling choices. Keeping in mind the current possibilities and limitations of XAI in drug discovery, it is reasonable to assume that the continued development of mixed approaches and alternative models that are more easily comprehensible and computationally affordable will not lose its importance.
At present, XAI in drug discovery lacks an open-community platform for sharing and improving software, model interpretations and the respective training data by synergistic efforts of researchers with different scientific backgrounds. Initiatives such as MELLODDY (Machine Learning Ledger Orchestration for Drug Discovery, melloddy.eu) for decentralized, federated model development and secure data handling across pharmaceutical companies constitute a first step in the right direction. Such kinds of collaboration will hopefully foster the development, validation and acceptance of XAI and the associated explanations these tools provide.
References
- Gawehn, E., Hiss, J. A. & Schneider, G. Deep learning in drug discovery. Mol. Inform.35, 3–14 (2016). Google Scholar
- Zhang, L., Tan, J., Han, D. & Zhu, H. From machine learning to deep learning: progress in machine intelligence for rational drug discovery. Drug Discov. Today22, 1680–1685 (2017). Google Scholar
- Muratov, E. N. et al. QSAR without borders. Chem. Soc. Rev.49, 3525–3564 (2020). Google Scholar
- Lenselink, E. B. et al. Beyond the hype: deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J. Cheminform.9, 45 (2017). Google Scholar
- Goh, G. B., Siegel, C., Vishnu, A., Hodas, N. O. & Baker, N. Chemception: a deep neural network with minimal chemistry knowledge matches the performance of expert-developed QSAR/QSPR models. Preprint at https://arxiv.org/abs/1706.06689 (2017).
- Unterthiner, T. et al. Deep learning as an opportunity in virtual screening. In Proc. Deep Learning Workshop at NIPS27, 1–9 (NIPS, 2014).
- Merk, D., Friedrich, L., Grisoni, F. & Schneider, G. De novo design of bioactive small molecules by artificial intelligence. Mol. Inform.37, 1700153 (2018). Google Scholar
- Zhavoronkov, A. et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol.37, 1038–1040 (2019). Google Scholar
- Schwaller, P., Gaudin, T., Lanyi, D., Bekas, C. & Laino, T. ‘Found in translation’: predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models. Chem. Sci.9, 6091–6098 (2018). Google Scholar
- Coley, C. W., Green, W. H. & Jensen, K. F. Machine learning in computer-aided synthesis planning. Acc. Chem. Res.51, 1281–1289 (2018). Google Scholar
- Senior, A. W. et al. Improved protein structure prediction using potentials from deep learning. Nature577, 706–710 (2020). Google Scholar
- Öztürk, H., Özgür, A. & Ozkirimli, E. DeepDTA: deep drug–target binding affinity prediction. Bioinformatics34, i821–i829 (2018). Google Scholar
- Jimenez, J. et al. Pathwaymap: molecular pathway association with self-normalizing neural networks. J. Chem. Inf. Model.59, 1172–1181 (2018). Google Scholar
- Marchese Robinson, R. L., Palczewska, A., Palczewski, J. & Kidley, N. Comparison of the predictive performance and interpretability of random forest and linear models on benchmark data sets. J. Chem. Inf. Model.57, 1773–1792 (2017). Google Scholar
- Webb, S. J., Hanser, T., Howlin, B., Krause, P. & Vessey, J. D. Feature combination networks for the interpretation of statistical machine learning models: application to Ames mutagenicity. J. Cheminform.6, 8 (2014). Google Scholar
- Grisoni, F., Consonni, V. & Ballabio, D. Machine learning consensus to predict the binding to the androgen receptor within the CoMPARA project. J. Chem. Inf. Model.59, 1839–1848 (2019). Google Scholar
- Chen, Y., Stork, C., Hirte, S. & Kirchmair, J. NP-scout: machine learning approach for the quantification and visualization of the natural product-likeness of small molecules. Biomolecules9, 43 (2019). Google Scholar
- Riniker, S. & Landrum, G. A. Similarity maps—a visualization strategy for molecular fingerprints and machine-learning methods. J. Cheminform.5, 43 (2013). Google Scholar
- Marcou, G. et al. Interpretability of sar/qsar models of any complexity by atomic contributions. Mol. Inform.31, 639–642 (2012). Google Scholar
- Rudin, C. Stop explaining black box machine learning models for high stakes decisions and use interpretable models instead. Nat. Mach. Intell.1, 206–215 (2019). Google Scholar
- Gupta, M., Lee, H. J., Barden, C. J. & Weaver, D. F. The blood–brain barrier (BBB) score. J. Med. Chem.62, 9824–9836 (2019). Google Scholar
- Rankovic, Z. CNS physicochemical property space shaped by a diverse set of molecules with experimentally determined exposure in the mouse brain: miniperspective. J. Med. Chem.60, 5943–5954 (2017). Google Scholar
- Leeson, P. D. & Young, R. J. Molecular property design: does everyone get it? ACS Med. Chem. Lett.6, 722–725 (2015). Google Scholar
- Fujita, T. & Winkler, D. A. Understanding the roles of the “two QSARs”. J. Chem. Inf. Model.56, 269–274 (2016). Google Scholar
- Schneider, P. et al. Rethinking drug design in the artificial intelligence era. Nat. Rev. Drug Discov.19, 353–364 (2020). Google Scholar
- Hirst, J. D., King, R. D. & Sternberg, M. J. Quantitative structure–activity relationships by neural networks and inductive logic programming. I. The inhibition of dihydrofolate reductase by pyrimidines. J. Comput. Aided Mol. Des.8, 405–420 (1994). Google Scholar
- Fiore, M., Sicurello, F. & Indorato, G. An integrated system to represent and manage medical knowledge. Medinfo.8, 931–933 (1995). Google Scholar
- Goebel, R. et al. Explainable AI: the new 42? In Machine Learning and Knowledge Extraction. CD-MAKE 2018. Lecture Notes in Computer Science Vol. 11015 (eds Holzinger, A., Kieseberg, P., Tjoa, A. & Weippl, E) (Springer, 2018).
- Lipton, Z. C. The mythos of model interpretability. Queue16, 31–57 (2018). Google Scholar
- Murdoch, W. J., Singh, C., Kumbier, K., Abbasi-Asl, R. & Yu, B. Definitions, methods, and applications in interpretable machine learning. Proc. Natl Acad. Sci. USA116, 22071–22080 (2019). MathSciNetMATHGoogle Scholar
- Doshi-Velez, F. & Kim, B. Towards a rigorous science of interpretable machine learning. Preprint at https://arxiv.org/abs/1702.08608 (2017).
- Lapuschkin, S. et al. Unmasking clever Hans predictors and assessing what machines really learn. Nat. Commun.10, 1096 (2019). Google Scholar
- Miller, T. Explanation in artificial intelligence: insights from the social sciences. Artif. Intell.267, 1–38 (2019). MathSciNetMATHGoogle Scholar
- Chander, A., Srinivasan, R., Chelian, S., Wang, J. & Uchino, K. Working with beliefs: AI transparency in the enterprise. In Joint Proceedings of the ACM IUI 2018 Workshops co-located with the 23rd ACM Conference on Intelligent User Interfaces 2068 (eds Said, A. & Komatsu, T.) (CEUR-WS.org, 2018).
- Guidotti, R. et al. A survey of methods for explaining black box models. ACM Comput. Surv.51, 93 (2018). Google Scholar
- Lundberg, S. M. et al. From local explanations to global understanding with explainable AI for trees. Nat. Mach. Intell.2, 2522–5839 (2020). Google Scholar
- Bendassolli, P. F. Theory building in qualitative research: reconsidering the problem of induction. Forum Qual. Soc. Res.14, 20 (2013). Google Scholar
- Schneider, P. & Schneider, G. De novo design at the edge of chaos: .iniperspective. J. Med. Chem.59, 4077–4086 (2016). Google Scholar
- Liao, Q. V., Gruen, D. & Miller, S. Questioning the AI: informing design practices for explainable AI user experiences. In Proc. 2020 CHI Conference on Human Factors in Computing Systems, CHI ‘20 1–15 (ACM, 2020).
- Sheridan, R. P. Interpretation of QSAR models by coloring atoms according to changes in predicted activity: how robust is it? J. Chem. Inf. Model.59, 1324–1337 (2019). Google Scholar
- Preuer, K., Klambauer, G., Rippmann, F., Hochreiter, S. & Unterthiner, T. in Interpretable Deep Learning in Drug Discovery (eds Samek W. et al.) 331–345 (Springer, 2019).
- Xu, Y., Pei, J. & Lai, L. Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J. Chem. Inf. Model.57, 2672–2685 (2017). Google Scholar
- Ciallella, H. L. & Zhu, H. Advancing computational toxicology in the big data era by artificial intelligence: data-driven and mechanism-driven modeling for chemical toxicity. Chem. Res. Toxicol.32, 536–547 (2019). Google Scholar
- Dey, S., Luo, H., Fokoue, A., Hu, J. & Zhang, P. Predicting adverse drug reactions through interpretable deep learning framework. BMC Bioinform.19, 476 (2018). Google Scholar
- Kutchukian, P. S. et al. Inside the mind of a medicinal chemist: the role of human bias in compound prioritization during drug discovery. PLoS ONE7, e48476 (2012). Google Scholar
- Boobier, S., Osbourn, A. & Mitchell, J. B. Can human experts predict solubility better than computers? J. Cheminform.9, 63 (2017). Google Scholar
- Sundararajan, M., Taly, A. & Yan, Q. Axiomatic attribution for deep networks. In Proc. 34th International Conference on Machine Learning Vol. 70, 3319–3328 (JMLR.org, 2017).
- Smilkov, D., Thorat, N., Kim, B., Viégas, F. & Wattenberg, M. Smoothgrad: Removing noise by adding noise. Preprint at https://arxiv.org/abs/1706.03825 (2017).
- Rumelhart, D. E., Hinton, G. E. & Williams, R. J. Learning representations by back-propagating errors. Nature323, 533–536 (1986). MATHGoogle Scholar
- Adebayo, J. et al. Sanity checks for saliency maps. Adv. Neural Inf. Processing. Syst .31, 9505–9515 (2018). Google Scholar
- Lipovetsky, S. & Conklin, M. Analysis of regression in game theory approach. Appl. Stoch. Models Bus. Ind.17, 319–330 (2001). MathSciNetMATHGoogle Scholar
- Lundberg, S. M. & Lee, S.-I. A unified approach to interpreting model predictions. Adv. Neural Inf. Process. Syst.30, 4765–4774 (2017). Google Scholar
- Ribeiro, M. T., Singh, S. & Guestrin, C. “Why should I trust you?” Explaining the predictions of any classifier. In Proc. 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining 1135–1144 (ACM, 2016).
- Shrikumar, A., Greenside, P. & Kundaje, A. Learning important features through propagating activation differences. In Proc. 34th International Conference on Machine Learning Vol. 70, 3145–3153 (JMLR.org, 2017).
- Bach, S. et al. On pixel-wise explanations for non-linear classifier decisions by layer-wise relevance propagation. PLoS ONE10, 1–46 (2015). Google Scholar
- Lakkaraju, H., Kamar, E., Caruana, R. & Leskovec, J. Interpretable & explorable approximations of black box models. Preprint at https://arxiv.org/abs/1707.01154 (2017).
- Deng, H. Interpreting tree ensembles with intrees. Int. J. Data Sci. Anal.7, 277–287 (2019). Google Scholar
- Bastani, O., Kim, C. & Bastani, H. Interpreting blackbox models via model extraction. Preprint at https://arxiv.org/abs/1705.08504 (2017).
- Maier, H. R. & Dandy, G. C. The use of artificial neural networks for the prediction of water quality parameters. Water Resour. Res.32, 1013–1022 (1996). Google Scholar
- Balls, G., Palmer-Brown, D. & Sanders, G. Investigating microclimatic influences on ozone injury in clover (Trifolium subterraneum) using artificial neural networks. New Phytol.132, 271–280 (1996). Google Scholar
- Štrumbelj, E., Kononenko, I. & Šikonja, M. R. Explaining instance classifications with interactions of subsets of feature values. Data Knowl. Eng.68, 886–904 (2009). Google Scholar
- Fong, R. C. & Vedaldi, A. Interpretable explanations of black boxes by meaningful perturbation. In Proc. IEEE International Conference on Computer Vision 3429–3437 (IEEE, 2017).
- Olden, J. D. & Jackson, D. A. Illuminating the “black box”: a randomization approach for understanding variable contributions in artificial neural networks. Ecol. Model.154, 135–150 (2002). Google Scholar
- Zintgraf, L. M., Cohen, T. S., Adel, T. & Welling, M. Visualizing deep neural network decisions: prediction difference analysis. Preprint at https://arxiv.org/abs/1702.04595 (2017).
- Ancona, M., Ceolini, E., Öztireli, C. & Gross, M. Towards better understanding of gradient-based attribution methods for deep neural networks. Preprint at https://arxiv.org/abs/1711.06104 (2017).
- McCloskey, K., Taly, A., Monti, F., Brenner, M. P. & Colwell, L. J. Using attribution to decode binding mechanism in neural network models for chemistry. Proc. Natl Acad. Sci. USA116, 11624–11629 (2019). Google Scholar
- Pope, P. E., Kolouri, S., Rostami, M., Martin, C. E. & Hoffmann, H. Explainability methods for graph convolutional neural networks. In Proc. IEEE Conference on Computer Vision and Pattern Recognition 10772–10781 (IEEE, 2019).
- Selvaraju, R. R. et al. Grad-cam: visual explanations from deep networks via gradient-based localization. In Pro. IEEE International Conference on Computer Vision 618–626 (IEEE, 2017).
- Zhang, J. et al. Top-down neural attention by excitation backprop. Int. J. Comput. Vis.126, 1084–1102 (2018). Google Scholar
- Tice, R. R., Austin, C. P., Kavlock, R. J. & Bucher, J. R. Improving the human hazard characterization of chemicals: a Tox21 update. Environ. Health Perspect.121, 756–765 (2013). Google Scholar
- Rodríguez-Pérez, R. & Bajorath, J. Interpretation of compound activity predictions from complex machine learning models using local approximations and Shapley values. J. Med. Chem.63, 8761–8777 (2019). Google Scholar
- Hochuli, J., Helbling, A., Skaist, T., Ragoza, M. & Koes, D. R. Visualizing convolutional neural network protein-ligand scoring. J. Mol. Graph. Model.84, 96–108 (2018). Google Scholar
- Jiménez-Luna, J., Skalic, M., Martinez-Rosell, G. & De Fabritiis, G. KDEEP: protein–ligand absolute binding affinity prediction via 3D-convolutional neural networks. J. Chem. Inf. Model.58, 287–296 (2018). Google Scholar
- Jiménez-Luna, J. et al. DeltaDelta neural networks for lead optimization of small molecule potency. Chem. Sci.10, 10911–10918 (2019). Google Scholar
- Todeschini, R. & Consonni, V. in Molecular Descriptors for Chemoinformatics: Volume I: Alphabetical Listing/Volume II: Appendices, References Vol. 41 (eds. Mannhold, R. et al.) 1–967 (Wiley, 2009).
- Hochreiter, S. & Schmidhuber, J. Long short-term memory. Neural Comput.9, 1735–1780 (1997). Google Scholar
- Vaswani, A. et al. Attention is all you need. Adv. Neural Inf. Process. Syst .30, 5998–6008 (2017). Google Scholar
- Weininger, D. Smiles, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci.28, 31–36 (1988). Google Scholar
- Grisoni, F. & Schneider, G. De novo molecular design with generative long short-term memory. CHIMIA Int. J. Chem.73, 1006–1011 (2019). Google Scholar
- Karpov, P., Godin, G. & Tetko, I. V. Transformer-CNN: Swiss knife for QSAR modeling and interpretation. J. Cheminform.12, 17 (2020). Google Scholar
- Doshi-Velez, F. et al. Accountability of AI under the law: the role of explanation. Preprint at https://arxiv.org/abs/1711.01134 (2017).
- Ribeiro, M. T., Singh, S. & Guestrin, C. Anchors: high-precision model-agnostic explanations. In Thirty-Second AAAI Conference on Artificial Intelligence 1527–1535 (AAAI, 2018).
- Wachter, S., Mittelstadt, B. & Russell, C. Counterfactual explanations without opening the black box: automated decisions and the GDPR. Harv. J. Law Technol.31, 841–888 (2017). Google Scholar
- Van Looveren, A. & Klaise, J. Interpretable counterfactual explanations guided by prototypes. Preprint at https://arxiv.org/abs/1907.02584 (2019).
- Dhurandhar, A. et al. Explanations based on the missing: towards contrastive explanations with pertinent negatives. Adv. Neural Inf. Process. Syst.31, 592–603 (2018). Google Scholar
- Zou, H. & Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B67, 301–320 (2005). MathSciNetMATHGoogle Scholar
- Mousavi, A., Dasarathy, G. & Baraniuk, R. G. Deepcodec: adaptive sensing and recovery via deep convolutional neural networks. Preprint at https://arxiv.org/abs/1707.03386 (2017).
- Randić, M., Brissey, G. M., Spencer, R. B. & Wilkins, C. L. Search for all self-avoiding paths for molecular graphs. Comput. Chem.3, 5–13 (1979). MATHGoogle Scholar
- Bonchev, D. & Trinajstić, N. Information theory, distance matrix, and molecular branching. J. Chem. Phys.67, 4517–4533 (1977). Google Scholar
- Duvenaud, D. K. et al. Convolutional networks on graphs for learning molecular fingerprints. Adv. Neural Inf. Process. Syst.28, 2224–2232 (2015). Google Scholar
- Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O. & Dahl, G. E. Neural message passing for quantum chemistry. In Proc. 34th International Conference on Machine Learning Vol. 70, 1263–1272 (JMLR.org, 2017).
- Krizhevsky, A., Sutskever, I. & Hinton, G. E. ImageNet classification with deep convolutional neural networks. Adv. Neural Inf. Process. Syst.25, 1097–1105 (2012). Google Scholar
- Kim, Y. Convolutional neural networks for sentence classification. Preprint at https://arxiv.org/abs/1408.5882 (2014).
- Kearnes, S., McCloskey, K., Berndl, M., Pande, V. & Riley, P. Molecular graph convolutions: moving beyond fingerprints. J. Comput. Aided Mol. Des.30, 595–608 (2016). Google Scholar
- Wu, Z. et al. Moleculenet: a benchmark for molecular machine learning. Chem. Sci.9, 513–530 (2018). Google Scholar
- Jin, W., Barzilay, R. & Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. Preprint at https://arxiv.org/abs/1802.04364 (2018).
- Baldassarre, F. & Azizpour, H. Explainability techniques for graph convolutional networks. In International Conference on Machine Learning (ICML) Workshops, 2019 Workshop on Learning and Reasoning with Graph-Structured Representations (ICML, 2019).
- Ying, Z., Bourgeois, D., You, J., Zitnik, M. & Leskovec, J. GNNExplainer: generating explanations for graph neural networks. Adv. Neural Inf. Process. Syst.32, 9240–9251 (2019). Google Scholar
- Veličković, P. et al. Graph attention networks. Preprint at https://arxiv.org/abs/1710.10903 (2017).
- Debnath, A. K., Lopez de Compadre, R. L., Debnath, G., Shusterman, A. J. & Hansch, C. Structure–activity relationship of mutagenic aromatic and heteroaromatic nitro compounds. Correlation with molecular orbital energies and hydrophobicity. J. Med. Chem.34, 786–797 (1991). Google Scholar
- Ishida, S., Terayama, K., Kojima, R., Takasu, K. & Okuno, Y. Prediction and interpretable visualization of retrosynthetic reactions using graph convolutional networks. J. Chem. Inf. Model.59, 5026–5033 (2019). Google Scholar
- Shang, C. et al. Edge attention-based multi-relational graph convolutional networks. Preprint at https://arxiv.org/abs/1802.04944 (2018).
- Ryu, S., Lim, J., Hong, S. H. & Kim, W. Y. Deeply learning molecular structure–property relationships using attention-and gate-augmented graph convolutional network. Preprint at https://arxiv.org/abs/1805.10988 (2018).
- Coley, C. W. et al. A graph-convolutional neural network model for the prediction of chemical reactivity. Chem. Sci.10, 370–377 (2019). Google Scholar
- Laugel, T., Lesot, M.-J., Marsala, C., Renard, X. & Detyniecki, M. The dangers of post-hoc interpretability: unjustified counterfactual explanations. In Proceedings of the 28th International Joint Conference on Artificial Intelligence 2801–2807 (AAAI, 2019)
- Melis, D. A. & Jaakkola, T. Towards robust interpretability with self-explaining neural networks. Adv. Neural Inf. Process. Syst.31, 7775–7784 (2018). Google Scholar
- Leake, D. B. in Case-based Reasoning: Experiences, Lessons and Future Directions, ch. 2 (ed. Leake, D. B.) (MIT Press, 1996).
- Kim, B., Rudin, C. & Shah, J. A. The Bayesian case model: a generative approach for case-based reasoning and prototype classification.Adv. Neural Inf. Process. Syst.27, 1952–1960 (2014). Google Scholar
- Li, O., Liu, H., Chen, C. & Rudin, C. Deep learning for case-based reasoning through prototypes: a neural network that explains its predictions. In Thirty-Second AAAI Conference on Artificial Intelligence 3530–3538 (AAAI, 2018).
- Chen, C. et al. This looks like that: deep learning for interpretable image recognition. Adv. Neural Inf. Process. Syst.32, 8928–8939 (2019). Google Scholar
- Goodman, N. D., Tenenbaum, J. B. & Gerstenberg, T. Concepts in a Probabilistic Language of Thought Technical Report (Center for Brains, Minds and Machines, 2014).
- Lake, B. M., Salakhutdinov, R. & Tenenbaum, J. B. Human-level concept learning through probabilistic program induction. Science350, 1332–1338 (2015). MathSciNetMATHGoogle Scholar
- Ghahramani, Z. Probabilistic machine learning and artificial intelligence. Nature521, 452–459 (2015). Google Scholar
- Vinyals, O., Blundell, C., Lillicrap, T., Kavukcuoglu, K. & Wierstra, D. Matching networks for one shot learning. Adv. Neural Inf. Process. Syst.29, 3630–3638 (2016). Google Scholar
- Altae-Tran, H., Ramsundar, B., Pappu, A. S. & Pande, V. Low-data drug discovery with one-shot learning. ACS Cent. Sci.3, 283–293 (2017). Google Scholar
- Kim, B., Wattenberg, M., Gilmer, J., Cai, C., Wexler, J., & Viegas, F. Interpretability beyond feature attribution: Quantitative testing with concept activation vectors (TCAV). In International Conference on Machine Learning 2668–2677 (2018).
- Gilpin, L. H. et al. Explaining explanations: an overview of interpretability of machine learning. In 2018 IEEE 5th International Conference on Data Science and Advanced Analytics (DSAA) 80–89 (IEEE, 2018).
- Hendricks, L. A. et al. Generating visual explanations. Computer Vision – ECCV 2016. ECCV 2016. Lecture Notes in Computer Science Vol. 9908 (eds Leibe, B., Matas, J., Sebe, N. & Welling, M.) (Springer, 2016).
- Antol, S. et al. VQA: visual question answering. In Proc. IEEE International Conference on Computer Vision 2425–2433 (IEEE, 2015).
- Rasmussen, C. E. Gaussian processes in machine learning. In Advanced Lectures on Machine Learning. Lecture Notes in Computer Science Vol. 3176 (Springer, 2004).
- Nguyen, A., Yosinski, J. & Clune, J. Deep neural networks are easily fooled: high confidence predictions for unrecognizable images. In Proc. IEEE Conference on Computer Vision and Pattern Recognition 427–436 (IEEE, 2015).
- Hansen, L. K. & Salamon, P. Neural network ensembles. IEEE Trans. Pattern Anal. Mach. Intell.12, 993–1001 (1990). Google Scholar
- Lakshminarayanan, B., Pritzel, A. & Blundell, C. Simple and scalable predictive uncertainty estimation using deep ensembles. Adv. Neural Inf. Process. Syst.30, 6402–6413 (2017). Google Scholar
- Freedman, D. A. Bootstrapping regression models. Ann. Stat.9, 1218–1228 (1981). MathSciNetMATHGoogle Scholar
- Huang, G. et al. Snapshot ensembles: train one, get m for free. Preprint at https://arxiv.org/abs/1704.00109 (2017).
- Zhang, R., Li, C., Zhang, J., Chen, C. & Wilson, A. G. Cyclical stochastic gradient MCMC for Bayesian deep learning. Preprint at https://arxiv.org/abs/1902.03932 (2019).
- Graves, A. Practical variational inference for neural networks. Adv. Neural Inf. Process. Syst.24, 2348–2356 (2011). Google Scholar
- Sun, S., Zhang, G., Shi, J. & Grosse, R. Functional variational bayesian neural networks. Preprint at https://arxiv.org/abs/1903.05779 (2019).
- Gal, Y. & Ghahramani, Z. Dropout as a bayesian approximation: representing model uncertainty in deep learning. In International Conference on Machine Learning 1050–1059 (JMLR, 2016).
- Kendall, A. & Gal, Y. What uncertainties do we need in bayesian deep learning for computer vision? Adv. Neural Inf. Process. Syst.30, 5574–5584 (2017). Google Scholar
- Teye, M., Azizpour, H., & Smith, K. Bayesian uncertainty estimation for batch normalized deep networks. In International Conference on Machine Learning 4907–4916 (2018).
- Nix, D. A. & Weigend, A. S. Estimating the mean and variance of the target probability distribution. In Proc. 1994 IEEE International Conference on Neural Networks (ICNN’94) Vol. 1, 55–60 (IEEE, 1994).
- Chryssolouris, G., Lee, M. & Ramsey, A. Confidence interval prediction for neural network models. IEEE Trans. Neural Netw.7, 229–232 (1996). Google Scholar
- Hwang, J. G. & Ding, A. A. Prediction intervals for artificial neural networks. J. Am. Stat. Assoc.92, 748–757 (1997). MathSciNetMATHGoogle Scholar
- Khosravi, A., Nahavandi, S., Creighton, D. & Atiya, A. F. Lower upper bound estimation method for construction of neural network-based prediction intervals. IEEE Trans. Neural Netw.22, 337–346 (2010). Google Scholar
- Ak, R., Vitelli, V. & Zio, E. An interval-valued neural network approach for uncertainty quantification in short-term wind speed prediction. IEEE Trans. Neural Netw. Learn. Syst.26, 2787–2800 (2015). MathSciNetGoogle Scholar
- Jiang, H., Kim, B., Guan, M. & Gupta, M. To trust or not to trust a classifier. Adv. Neural Inf. Process. Syst.31, 5541–5552 (2018). Google Scholar
- Huang, W., Zhao, D., Sun, F., Liu, H. & Chang, E. Scalable Gaussian process regression using deep neural networks. In Twenty-Fourth International Joint Conference on Artificial Intelligence 3576–3582 (AAAI, 2015).
- Sheridan, R. P., Feuston, B. P., Maiorov, V. N. & Kearsley, S. K. Similarity to molecules in the training set is a good discriminator for prediction accuracy in QSAR. J. Chem. Inf. Comput. Sci.44, 1912–1928 (2004). Google Scholar
- Liu, R. & Wallqvist, A. Molecular similarity-based domain applicability metric efficiently identifies out-of-domain compounds. J. Chem. Inf. Model.59, 181–189 (2018). Google Scholar
- Janet, J. P., Duan, C., Yang, T., Nandy, A. & Kulik, H. J. A quantitative uncertainty metric controls error in neural network-driven chemical discovery. Chemi. Sci.10, 7913–7922 (2019). Google Scholar
- Scalia, G., Grambow, C. A., Pernici, B., Li, Y.-P. & Green, W. H. Evaluating scalable uncertainty estimation methods for deep learning-based molecular property prediction. J. Chem. Inf. Model.60, 2697–2717 (2020). Google Scholar
- Obrezanova, O., Csányi, G., Gola, J. M. & Segall, M. D. Gaussian processes: a method for automatic QSAR modeling of ADME properties. J. Chem. Inf. Model.47, 1847–1857 (2007). Google Scholar
- Schroeter, T. S. et al. Estimating the domain of applicability for machine learning QSAR models: a study on aqueous solubility of drug discovery molecules. J. Comput. Aided Mol. Des.21, 485–498 (2007). Google Scholar
- Bosc, N. et al. Large scale comparison of QSAR and conformal prediction methods and their applications in drug discovery. J. Cheminform.11, 4 (2019). Google Scholar
- Cortés-Ciriano, I. & Bender, A. Deep confidence: a computationally efficient framework for calculating reliable prediction errors for deep neural networks. J. Chem. Inf. Model.59, 1269–1281 (2018). Google Scholar
- Schwaller, P. et al. Molecular transformer: a model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci.5, 1572–1583 (2019). Google Scholar
- Zhang, Y. & Lee, A. A. Bayesian semi-supervised learning for uncertainty-calibrated prediction of molecular properties and active learning. Chem. Sci.10, 8154–8163 (2019). Google Scholar
- Hirschfeld, L., Swanson, K., Yang, K., Barzilay, R. & Coley, C. W. Uncertainty quantification using neural networks for molecular property prediction. Preprint at https://arxiv.org/abs/2005.10036 (2020).
- Kokhlikyan, N. et al. PyTorch Captum. GitHubhttps://github.com/pytorch/captum (2019).
- Paszke, A. et al. PyTorch: an imperative style, high-performance deep learning library. Adv. Neural Inf. Process. Syst.32, 8026–8037 (2019). Google Scholar
- Klaise, J., Van Looveren, A., Vacanti, G. & Coca, A. Alibi: algorithms for monitoring and explaining machine learning models. GitHubhttps://github.com/SeldonIO/alibi (2020).
- Pedregosa, F. et al. Scikit-learn: machine learning in Python. J. Mach. Learn. Res.12, 2825–2830 (2011). MathSciNetMATHGoogle Scholar
- Abadi, M. et al. TensorFlow: a system for large-scale machine learning. In 12th USENIX Symposium on Operating Systems Design and Implementation (OSDI 16) 265–283 (USENIX Association, 2016).
- Lipton, Z. C. The doctor just won’t accept that! Preprint at https://arxiv.org/abs/1711.08037 (2017).
- Goodman, B. & Flaxman, S. European Union regulations on algorithmic decision-making and a ‘right to explanation’. AI Mag.38, 50–57 (2017). Google Scholar
- Ikebata, H., Hongo, K., Isomura, T., Maezono, R. & Yoshida, R. Bayesian molecular design with a chemical language model. J. Comput. Aided Mol. Des.31, 379–391 (2017). Google Scholar
- Segler, M. H., Kogej, T., Tyrchan, C. & Waller, M. P. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent. Sci.4, 120–131 (2018). Google Scholar
- Nagarajan, D. et al. Computational antimicrobial peptide design and evaluation against multidrug-resistant clinical isolates of bacteria. J. Biol. Chem.293, 3492–3509 (2018). Google Scholar
- Müller, A. T., Hiss, J. A. & Schneider, G. Recurrent neural network model for constructive peptide design. J. Chem. Inf. Model.58, 472–479 (2018). Google Scholar
- Jiménez-Luna, J., Cuzzolin, A., Bolcato, G., Sturlese, M. & Moro, S. A deep-learning approach toward rational molecular docking protocol selection. Molecules25, 2487 (2020). Google Scholar
- Rogers, D. & Hahn, M. Extended-connectivity fingerprints. J. Chem. Inf. Model.50, 742–754 (2010). Google Scholar
- Awale, M. & Reymond, J.-L. Atom pair 2D-fingerprints perceive 3D-molecular shape and pharmacophores for very fast virtual screening of ZINC and GDB-17. J. Chem. Inf. Model.54, 1892–1907 (2014). Google Scholar
- Todeschini, R. & Consonni, V. New local vertex invariants and molecular descriptors based on functions of the vertex degrees. MATCH Commun. Math. Comput. Chem.64, 359–372 (2010). MathSciNetGoogle Scholar
- Katritzky, A. R. & Gordeeva, E. V. Traditional topological indexes vs electronic, geometrical, and combined molecular descriptors in QSAR/QSPR research. J. Chem. Inf. Comput. Sci.33, 835–857 (1993). Google Scholar
- Sahigara, F. et al. Comparison of different approaches to define the applicability domain of qsar models. Molecules17, 4791–4810 (2012). Google Scholar
- Mathea, M., Klingspohn, W. & Baumann, K. Chemoinformatic classification methods and their applicability domain. Mol. Inform.35, 160–180 (2016). Google Scholar
- Liu, R., Wang, H., Glover, K. P., Feasel, M. G. & Wallqvist, A. Dissecting machine-learning prediction of molecular activity: is an applicability domain needed for quantitative structure–activity relationship models based on deep neural networks? J. Chem. Inf. Model.59, 117–126 (2019). Google Scholar
- Nembri, S., Grisoni, F., Consonni, V. & Todeschini, R. In silico prediction of cytochrome P450-drug interaction: QSARs for CYP3A4 and CYP2C9. Int. J. Mol. Sci.17, 914 (2016). Google Scholar
- Waller, D., Renwick, A., Gruchy, B. & George, C. The first pass metabolism of nifedipine in man. Br. J. Clin. Pharmacol.18, 951–954 (1984). Google Scholar
- Hiratsuka, M. et al. Characterization of human cytochrome p450 enzymes involved in the metabolism of cilostazol. Drug Metab. Dispos.35, 1730–1732 (2007). Google Scholar
- Raemsch, K. D. & Sommer, J. Pharmacokinetics and metabolism of nifedipine. Hypertension5, II18 (1983). Google Scholar
Acknowledgements
We thank N. Weskamp and P. Schneider for helpful feedback on the manuscript. This work was financially supported by the ETH RETHINK initiative, the Swiss National Science Foundation (grant no. 205321_182176) and Boehringer Ingelheim Pharma GmbH & Co. KG.
